-
MolTrans: Molecular Interaction Transformer for drug-target interaction predictiAI/Bio & Medical 2022. 11. 29. 17:49
MolTrans: Molecular Interaction Transformer for drug-target interaction prediction
Kexin Huang 1, Cao Xiao 2, Lucas M. Glass 2 and Jimeng Sun 3,*
MolTrans는 drug-target interaction (DTI)를 하기 위해서 Transformer를 적용한 논문이다.
healthcare 분야의 중요도가 높아지며 bio 분야에서도 AI를 활용한 활발한 연구가 진행되고 있다.
기존 DTI에서는 많은 시간과 비용이 필요했지만 AI의 도입과 함께 시간과 비용이 많이 줄어들게 되었다. 이렇듯 AI는 바이오 분야에서도 중요한 요소로 자리 잡게 되었다.
논문 doi : https://doi.org/10.1093/bioinformatics/btaa880https://academic.oup.com/bioinformatics/article/37/6/830/5929692
academic.oup.com
1. Introduction
Drug discovery는 over large drug compound space (대규모 약물 화합물 공간)에 대한 실험이 필요하기 때문에 많은 비용과 시간이 소요되는 것으로 악명이 높다.
그중에서 Drug-target protein interaction (DTI)는 약물 화합물의 치료효과를 감지하여 알맞은 반응의 약물을 찾는 행위로 신약 및 기존 약물의 새로운 적응증을 찾는데 기초가 된다.
이러한 화합물 identification process 동안 연구원은 분석 실험을 수행하고 후보 database에서 97M 이상의 possibile compounds를 찾아야 한다.
이렇듯 병변에 적합한 반응을 하는 화합물을 찾는 것에서 엄청난 비용과 많은 시간이 소요된다.
하지만 현재 많은 응용 분야에서 큰 성공을 거둔 deep learning 기술의 발전과 함께 방대한 생물 의학 데이터와 지식이 수집되고 이용 가능해짐에 따라 drug discovery process, 특히 DTI prediction 은 크게 발전하였다.
최근 다양한 deep models 가 DTI prediction에서 고무적인 성능을 보여주었다.
drug와 protein data를 입력으로 사용하여 DTI를 classification problem으로 casting 하고 deep neural network (DNN), deep belief network (DBN), convolutional neural network (CNN) 등을 사용하여 DTI prediction을 진행해왔었다.
그러나 이러한 연구에도 다음과 같은 문제들은 여전히 존재한다.
Problem 1 : Interaction mechanism에 부적절한 modeling
기존 연구는 molecular representation을 배우고 drug와 protein의 전체 분자 구조를 기반으로 예측한다. 그러나 이러한 방법은 intercations 가 drug와 protein의 하위 구조만 포함한다는 사실을 무시하게 된다.
이렇듯 전체 구조를 사용하게 되면 noise가 생기게 되고 예측 성능에 영향을 미치게 된다. 또한, 이렇게 학습된 representation 은 drug과 protein 의 어떤 하위 구조가 interaction에 기여하였는지에 대해서 다루기 쉬운 path를 제공하지 않기 때문에 해석하기가 어렵다.Problem 2 : Restricte dto limited labeled data
기존 연구는 현재 보유하고 있는 데이터에 초점을 맞추고 범위를 수천 개의 drug 및 protein 내로 제한하는 동시에 방대한 양의 label이 지정되지 않은 사용 가능한 biomedical data를 무시한다.
이전 연구의 모델 아키텍처는 또한 대구모 데이터 세트의 통합을 가능하게 하도록 설계되지 않았다.
이러한 문제를 해결하기 위해서 본 저자들은 Transformer 기반의 bio-inspired molecular data representation method [coined as Molecular Innteraction Transformer (MolTrans)] 를 제안한다. MolTrans의 contribution 은 다음과 같다.
Contribuition 1 : Knowledge inspired representation and interaction modeling for more accurrate and explainable prediction
DTI가 sub-structural이라는 지식에서 영감을 받은 MolTrans는 protein과 drug 모두에 대해 고품질의 적합한 크기의 sub-structural을 추출하는데 적용할 수 있는 Frequent Consecutive Sub-sequence(FCS) mining이라는 data-driven method를 사용한다.
또한, MolTrans 에는 실제 biological DTI process를 모방한 bio-inspired interaction module이 포함되어 있다. 새로운 sub-structure fingerprints를 사용하려면 interaction module의 explicit map을 통해 어떤 sub-structure 조합이 결과와 더 관련이 있는지 이해할 수 있는 다루기 쉬운 path를 사용할 수 있다.Contribution 2 : Leverage massive unlabeled biomedical data
MolTrans는 여러 un-labeled data sources에서 수백만 개의 drug 및 protein 서열을 조사하여 drug 및 protein의 high-quality sub-structures를 추출한다.
방대한 데이터는 소규모 train dataset 만 사용하는 것보다 훨씬 더 높은 품질의 sub-structures (하위 구조)를 생성한다. 또 un-labeled data에서 생성된 large sequential sub-structures outputs 중에서 복잡한 signal을 captures 하는 transformer를 사용하여 representation을 보강한다.
이들은 unseen drug/target problems 및 부족한 training dataset 설정을 포함 하여 다양한 현실적인 drug discovery setting에 대한 state-of-the-art method (sota) 들과의 성능 비교를 제공하고 있다. 또한 MolTrans가 sota에 비해 prediction 성능을 최대 25% 까지 높였음을 실험을 통해서 보여주고 있다.
2. Materials and methods
2.1 Problem definition
한 쌍의 drug와 target protein 이 상호 작용할지 여부를 결정하기 위해 DTI prediction을 classification problem으로 공식화한다.
Drug는 Simplified Molecular Input Line Entry System(SMILES) S_i로 표현된다. 이는 분자 그래프에 대한 depth-first traversal (DFT)에 의해 생선 된 일련의 chemical atoms 및 결합 토큰 (e.g. C, O, S)으로 구성된다.
또한 S는 drug's SMILES 표현을 뜻한다.
A로 표시된 target protein은 protein tokens의 sequence로 표시되면 각 token 은 23개의 amino acids 중 하나이다.
DTI prediction 은 아래와 같이 정의된다.Problem 1 : DTI Prediction
n drugs에 대한 compound sequence S = {S_1,..., S_n}
m proteins에 대한 protein sequence A = {A_1,..., A_m}
이 주어지면 DTI prediction 작업은 다음과 같이 casted 될 수 있다.
function mapping F: S x A -> [0, 1] from drug-target pairs to an interaction probability score.
2.2 The MolTrans method
이 section에서는 MolTrans에 대해서 소개한다.
입력 drug 및 protein data가 주어지면 FCS mining module 은 먼저 specialized decomposition algorithm을 사용하여 이를 sub-structures의 explicit sequences 세트로 분해한다.
그런 다음 outputs 은 transformer encoder를 통해 각 sub-structures에 대한 augmented contextual embedding을 얻기 위해서 augmented transformer embedding module의 input으로 사용된다.
다음으로, interaction prediction module에서 drug sub-structures는 interaction score 쌍으로 protein sub-structures와 쌍을 이룬다.
CNN layer는 나중에 interaction map에 적용되어 higher-order interactions를 capture 한다. 마지막으로, decoder module 은 쌍별 interaction의 확률을 나타내는 점수를 출력한다.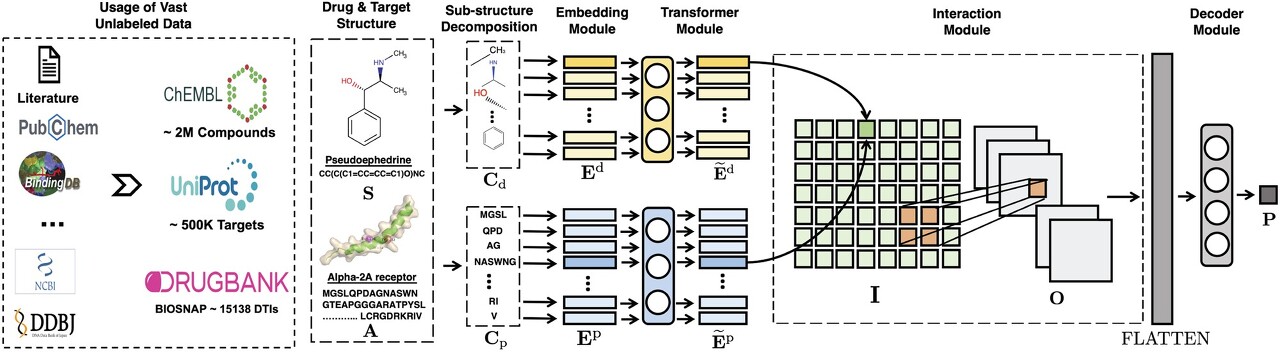
MolTrans의 아키텍쳐 2.2.1 FCS mining module
DTI가 sub-structural level에서 발생한다는 domain 지식을 기반으로 MolTrans는 먼저 protein 및 drug의 분자 서열을 sub-structures로 분해한다. 특히 drug 및 protein database에서 반복되는 하위 서열을 찾기 위해서 본 논문의 저자는 FCS algorithm이라는 데이터 기반 순차 패턴 마이닝 알고리즘 (data-driven sequential pattern-mining algorithm)을 제안한다.
NLP 분야의 invention of sub-word units에서 영감을 얻은 FCS는 sequence에 frequent sub-sequences의 계층 구조 집합을 생성하는 것을 목표로 한다. 다음 Algorithm 1은 FCS를 간단하게 표현한 것이다.
본 논문에서 제시된 FCS Algorithm FCS는 계층적으로 protein / drug 의 각 sequence를 하위 squence, 더 작은 하위 sequence 및 개별 atom/amino acids 기호로 분해한다.
FCS는 먼저 고유한 amino acids toekn(아미노산 토큰) 또는 SMILES 문자열 문자의 voclvulary set(어휘 세트) V를 초기화하고 token이 주어지면 전체 drug/protein corpus를 토큰화 한다.
토큰화 된 set를 W라고 부른다.
그런 다음, W를 통해 스캔하고 가장 빈번한 연속 토큰 (A, B)를 식별한다.
그런 다음 FCS는 토큰화 된 세트 W의 모든 (A, B)를 새 토큰 (AB)으로 업데이트하고 이 새 토큰을 어휘 세트 V에 추가한다.
이 스캔, 식별, 업데이트 process는 임계가 θ 를 초과하는 빈도가 높은 토큰이 없거나 V의 크기가 미리 정의돈 최댓값 l에 도달할 때까지 반복된다.
이 작업을 통해 빈도가 높은 하위 시퀀스는 하나의 토큰으로 병합되고 빈도가 낮은 하위 시퀀스는 더 짧은 토큰 집합으로 분해된다.
결국 drug/protein의 경우, FCS 는 크기가 k 인 하위 구조 drug/target protein의 서열 C = {C_1,..., C_k}를 생성하며, 여기서 C_i는 세트 V에서 나온다.
FCS algorithm을 사용하여 MolTrans는 입력 drug와 target을 각각 explicit sub-structures (명시적 하위 구조) C_d 및 C_p의 sequence로 변환한다. FCS의 중요성은 다음 세 가지 있다.- 더 설명하기 쉽다는 점에서 이전 sub-structure fingerprinting methods과 구별된다. PubChem encoding과 같은 명시적 하위 구조 fingerprint 에는 작은 분자에 대해 평균 100개의 세분화된 하위 구조가 있으며 많은 하위 구조가 다른 하위 구조의 집합이므로 어떤 하위 구조가 결과로 이어지는지 알기 어렵다. 반면, FCS drug encoding 은 각 drug molecular를 개별적이고 적당한 크기의 하위 구조 분할로 분해하므로 명시적인 힌트를 제공할 수 있다.
- 향상된 sub-structure mining을 위해 방대한 양의 un-labeled data를 활용할 수 있다. 작은 데이터 세트를 사용하는 것과 비교하여 대규모 un-labeled data를 사용할 때 encoding이 더 좋은 예측력을 갖는다는 것을 실험적으로 확인할 수 있다.
- FCS는 기본적이고 의미 있는 biomedical semantics를 포착할 수 있다. 생성된 하위 구조는 자주 반복되는 drug 및 protein의 기본 단위와 연결된다.
2.2.1 Augmented transformer embedding module
원래 transforemr는 self-attention 메커니즘을 활용하여 context embedding을 생성하는 state-of-the-art depp learning 아키텍처이며 자연어 처리 작업을 위해서 만들어졌다. 이를 해당 task에 맞는 모델로 만들기 위해서 molecue representation에 맞게 조정하여 학습시켰다.
이들의 setting에서 transformer encoder의 self-attention 메커니즘은 동일한 분자의 모든 sub-structures에서 학습하여 각 input sub-structures embedding을 수정하게 된다.
최종 sub-structural embedding 은 인접한 sub-structures 간의 복잡한 화학적 관계를 고려하기 때문에 더 좋은 sub-structural embedding 이 된다.
구체적으로 설명해 보자.
각 input drug-target pair에 대해서 sub-structures C_p 및 C_d의 해당 sequence를 두 개의 행렬 $$ M^{p}\in R^{k\times \theta _{p}}, M^{d}\in R^{l\times \theta _{d}} $$ 로 변환한다.
연기서 k/l 은 drug/protein 에 대한 sub-structures의 전체 크기 또는 FCS algorithm의 어휘 세트 V의 cardinality이고, $$ \theta _{p}, \theta _{d} $$ 는 protein 및 drug에 대한 sub-structures sequences의 최대 길이이며 $$ M^{p}_{i}, M^{d}_{j} $$ 는 protein sequence의 i번째 sub-structure 및 durg sequence의 j번째 sub-structure에 대한 sub-structure index에 해당하는 one-hot vector이다.
각 protein 및 drug에 대한
$$ E^{p}_{cont_{i} } $$
$$ E^{d}_{cont_{j} } $$
를 포함하는 content embedding 은 학습 가능한 dictionary lookup matric 인
$$ W^{p}_{cont} \in \mathbb {R}^{\vartheta \times k}$$
$$ W^{d}_{cont} \in \mathbb{R}^{\vartheta \times l} $$
을 통해서 생성되어 다음과 같은 수식을 얻을 수 있다.
이때 $$ \vartheta $$ 는 각 sub-structure에 대한 latent embedding의 size이다.
MolTrans는 sequential sub-structures (순차 하위 구조)를 사용하기 때문에 lookup dictionary
lookup dictionary 를 통해서 positional embedding

positional embedding 를 얻게 된다.
이를 수식으로 표현하면 다음과 같은 식을 얻을 수 있다.
position embedding 수식 이때,

은 i/j 번째 위치가 1인 single hot vector이다.
최종적으로 final embedding
final embedding 는 content 및 position embedding의 합을 통해서 생성된다.

final embedding 수식 위의 모델은 독립적인 sub-structure embedding set를 출력한다.
그러나 이러한 sub-structure는 context 정보를 캡처하기 위한 그 데이터들 사이의 Octet rules와 같은 화학적 관계가 있다. 이를 위해 transformer encoder layer를 사용하여 embedding을 추가로 확장해준다.
2.2.3 Interaction prediction module
MolTrans 에는 2-layer로 구성된 interaction module 이 포함되어 있다.
- pairwise sub-structural interaction을 modeling 하기 위한 interaction tensor
- neighborhood interaction (이웃 상호작용)을 추출하기 위한 interaction map 위의 CNN layer
2.2.3.1 Pairwise interaction
Pairwise interaction을 modeling 하기 위해 protein의 각 sub-sequence i와 drug의 sub-sequence j에 대해서 다음과 같은 식이 있다.

여기서 F는 pair 간의 상호 작용을 측정하는 함수이며 이 함수는 합계, 평균 및 내적과 같은 모든 함수가 될 수 있다.
따라서 이 layer 뒤에는 다음과 같은 tensor가 존재한다.
여기서..

은 각각 drug/protein 에 대한 sub-sequence의 길이이고

은 함수 F의 output size이다.
여기서 이 tensor의 각 열은 protein과 drug의 개별 sub-structure의 interaction (상호작용)을 고려한다.
보통 설명 가능성을 위해서 개별 targ-drug sub-structural pair 간의 상호 작용 강도를 명시적으로 측정하는 단일 스칼라를 생성하기 때문에 aggregation function으로는 내적 함수를 선호한다.
내적의 output 은 모든 쌍에 대해 1차원 이므로 I는 2차원 interaction map 이 된다.
map의 값이 높으면 downstream layer에서 활성화되어 DTI interaction 가능성이 높아진다.
end-to-end 학습을 통해 한 쌍의 sub-structure 가 실제로 상호 작용하면 interaction map에서 해당 sub-structure 위치에서 높은 interaction score를 갖게 된다.
따라서 이 map을 검토하여 어떤 sub-structure 쌍이 최종 결과에 더 기여하는지 직접 확인할 수 있다.
즉, 해당 수식을 통해서 얻은 값을 통해서 어떠한 하위 구조가 반응을 더 많이 일으키는지를 결정하게 되는 것이다.
2.2.3.2 Neighborhood interaction
protein과 drug의 nearby sub-structure (인접 하위 구조)는 interaction을 유발하는 데 서로 양향을 미친다.
따라서 individual pairwise interaction 을interaction을 modelinng 하는 것 외에도 nearby regions에 대한 interaction을 modeling 하는 것도 필요하다.
interaction map I 위에 있는 CNN layer를 통해서 이를 얻을 수 있다.
Convolution filter를 통해 nearby regions에 대한 interaction을 capture 하고 aggregated 할 수 있다는 것이다.
Drug-target pair를 input으로 사용하여 다음과 같이 output O를 얻을 수 있다.
이러한 interaction module 은 Deep Interactive Inferencee Network에서 영감을 받았다고 한다.
이 explicit interaction modeling 덕북에 나중에 interaction map에서 개별 sub-structural pair의 강도를 시각화할 수 있다.
interaction 가능성을 나타내는 확률을 출력하기 위해 먼저 O를 벡터로 flatten 하고 weight matrix $$ W_{o} $$ 및 bias vector $$ b_{o} $$ 를 parameterized 된 linear layer를 사용한다. 이를 통해 다음과 같은 수식을 얻을 수 있게 된다.
이때

이다.
다음 같은 paramters
network parameters 와 transformer encoder weights, CNN weights 가 있는 전체 network는 binary classification loss를 통해 최적화할 수 있다.
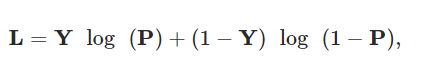
Binary classification loss 이때 Y는 ground truth label을 의미한다.
2.3 Implementation
본 논문에 저장들은 Pytorch를 이용하여 MolTrans를 구현하였다.
FCS algorithm의 경우 dataset에서 sub-structures의 최소 발생 수를 drug 및 protein에 대해 500으로 설정하여 23,532개의 drug sub-structures와 16,693개의 protein sub-structures 를 생성하였다.
Transformer encoder의 경우 drug와 protein 모두에 transformer encoder에 있는 두 개의 layer를 사용하였다.
input embedding size는 384이고 intermediate dimension이 1536인 각 transformer encoder에 대해 head를 12로 설정하여 multi-head attention을 진행하였다.
dataset에서 drug의 max sequence length를 50, protein의 max sequence length를 545로 설정하여 dataset의 95%를 커버하였다고 한다.
최대 길이의 이상/미만의 부분에 대해 cut/padding을 진행하였다.
CNN의 경우 kernel size가 3인 filter 3개를 사용한다.
optimizer로 Adam을 사용하였으며 learning rate는 1e-5로 설정하였다.
batch size를 64로 설정하고 epoch 는 30으로 설정하였다.
8~15 epoch 사이에 수렴함을 확인하였다고 한다.
또한 dropout rate는 0.1이다.input embedding size 384 intermediate dimension 1536 head 12 MAX sequence length of drug 50 MAX sequence length of protein 545 CNN kernel size / filters 3 x 3 / 3 Optimizer Adam Learning rate 1e-5 epoch 30 batch size 64 dropout 0.1
3. Result
이들은 binary classification의 성능을 측정하기 위해서 metrics로 ROC-AUC (area under the receiver operating characteristic curve)와 PR-AUC (are under the precision-recall curve)를 사용하였다.
또한 threshold가 validation set에서 가장 좋은 F1 score를 가진 것인 sensitivity와 specificity를 사용한다.
이들은 dataset을 7:1:2의 비율로 training, validation and testinng sets로 나누었다.
모든 실험에서 데이터 세트의 서로 다른 random split을 사용하여 5개의 독립적인 실행을 수행하였다.
Table 1. Dataset 위 table은 이들이 실험에 사용한 dataset을 정리한 것이다.

위 표는 각각에 dataset에 대하여 기존 model과 이들이 제안하는 모델의 성능을 비교한 것이다.
전체적으로 MolTrans의 성능이 더 높다는 것을 확인할 수 있다.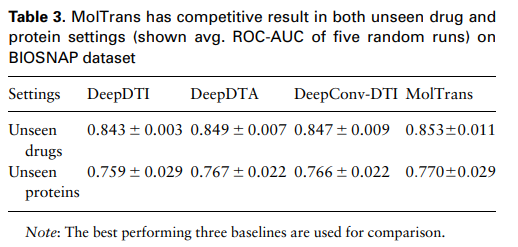
위 표는 Unseen drug/protein에 대한 성능을 나타낸 표이다. BIOSNAP dataset을 사용하였다고 한다.
성능이 모든 setting에서 MolTrans의 성능이 가장 우수함을 알 수 있다.
DTI 데이터의 가용성이 폭발적으로 증가하고 있지만 일부 실제 약물 발견 파이프라인에는 예산 제한으로 인해 소수의 label만 있는 새로운 target protein 또는 drug만 가지고 있는 경우가 있다.
그렇기 때문에 low resource 상황에서도 강력한 성능을 가지는 것이 가장 이상적인 DTI이다.
위 표는 low resource로 제약을 걸고 강력한 성능을 내는지를 확인하기 위해서 진행한 실험이다.
각 training set의 비율을 5%, 10%, 20%, 30% 로 하여 학습하고 나머지 data를 test 하였다.
이를 통해 low resource 제약에서도 MolTrans가 가장 우수한 성능을 가짐을 확인할 수 있다.
이는 MolTrans가 전체 durg와 protein을 활용하는 다른 방법에 비해 상대적으로 풍부한 sub-structures의 embedding을 활용하기 때문이다.
다음 실험은 CNN, transformer 및 interaction module이 최종 성능에 영향을 미치는 정도를 확인할 수 있는 실험이다.
FCS fingerprint 만으로도 interaction에서 강력한 성능을 보인다.
또한 Small에서는 방대한 un-labeld data가 input을 풍부하게 하고 성능을 향상하므로 유용하다는 것을 확인할 수 있다.
마치며..
본 논문은 drug target interaction을 위하여 transformer를 적용한 논문이다.
본래 DTI 작업에만 몇 년이라는 시간이 걸리지만 AI의 적용을 통해서 엄청나게 많은 시간이 단축되어 몇 개월이면 이를 분석할 수 있게 되었다.
인공지능은 현재 신약 개발 분야에서도 아주 큰 영향을 미치고 있다고 생각하며 앞으로 중요한 부분으로 자리 잡을 것이라 생각한다.
Domain에 대한 지식이 많지 않아 논문을 읽고 분석하는데 오랜 시간이 걸린 편이기에 이 분야로 공부를 계속하기 위해서는 domain공부를 함께 진행해야 할 필요가 있을 것 같다.
'AI > Bio & Medical' 카테고리의 다른 글