-
AlphaFold: Improved protein structure prediction using potentials form deep learningAI/Bio & Medical 2023. 2. 6. 02:38
NATURE
Improved protein structure prediction using potentials form deep learning
Google DeepMind
이번에 리뷰할 논문은 Improved protein structure prediction using potentials form deep learning이다.
AlphaFold라고 불리는 model을 제안한 논문이다.
CASP(Critical Assessment of Structure Prediction) 대회에서 1등을 차지하였으며 이후 나온 AlphaFold 2는 압도적인 성능차이로 작년 대회에서 1등을 차지하였다.
AlphaFold 2 를 리뷰하기 전 우선 AlphaFold 1에 대해서 알아보자.
doi : 10.1038/s41586-019-1923-7
0. Background
본 논문을 읽어보기전 우리는 단백질 구조가 왜 중요한지에 대해서 알아야 할 필요가 있다.
우선 질병의 원인은 크게 두 가지로 나누어서 볼 수 있다.
- 단백질 이상
- 외부의 미생물 침입
이에 대한 치료법으로는 크게 단백질 이상일 경우 해당 단백질의 기능을 정상화하거나 미생물의 침입일 경우 미생물의 필수 단백질의 기능을 막는 약을 섭취는 하는 것이 있다.
결국 질병을 치료하기 위한 약은 특정한 단백질을 타겟으로 잡고 진행하게 된다.

보통 약을 개발하는 분야에서는 {자물쇠: 단백질, 약: 열쇠} 라고 표현을 한다.
단백질은 특정한 3차원 구조를 지니고 있기 때문에, 이것에 딱 맞는 물질을 찾아낼 수 있다.
만약, 그 단백질이 질병과 연관이 되어 있다면, 이 단백질에 꼭 맞는 물질은 그 병의 치료제가 될 수 있을 것이다.
그럼 기존에 약 개발은 어떻게 진행되었는지 확인해 보자.
- 과거 : 경험적, 추상적 개념
- ex) 어떤 식물을 먹으니까, 고통이 줄어들더라!
- 현재 : 타겟 단백질의 구조 또는 약의 구조를 기반
- 타겟 단백질의 구조를 알고 접근
- 효능이 있는 구조를 알고 접근
그러나 현재의 개발은 자물쇠(단백질)의 모양을 모르고 다양한 열쇠(약)를 끼워 보는 방식이라고 얘기할 수 있다고 한다.
그렇기 때문에 새로운 약이 개발되는 기간이 너무 길고 많은 비용을 필요로 하게 된다.
단백질의 구조를 알 수만 있다면 약 개발의 기간과 비용을 효과적으로 줄일 수 있을 것이다.
1. Problem Definition
단백질 (Protein) 이란?
단백질은 아미노산이 사슬처럼 연결되어 만들어진 거대하고 복잡한 분자 구조이다.
단백질이 어떤 역할을 할지는 바로 각 아미노산의 구조에 달려있다.
아미노산 사슬은 아미노산이 어떻게 접히는지가 아닌 아미노산의 순서에 대한 정보만을 내포하기 때문에, 단백질의 기본 단위인 아미노산이 어떻게 입체적으로 배열되고 접혀 3차원 구조의 단백질을 형성하는지 연구해야 할 필요가 있다.
어떤 단백질의 아미노산 사슬이 주어졌을 때, 그 단백질의 3차원 구조를 예측하는 단백질 접힘(Protein Folding) 문제는 수십 년간 과학자들이 해결하고자 했던 난제 중 하나였다고 한다.
단백질 접힘 문제는 왜 중요한 걸까?

https://spectrum.ieee.org/gpt-2-language-model-proteins 단백질의 접힘에 의해 만들어진 고유한 구조는 단백질의 기능과 직결되기 때문이다.
그래서 단백질이 잘못 접히면 다양한 질환과 연관될 수 있다.
단백질 잘못 접힘은 단백질이 고유한 3차원 구조를 형성하지 못하게 되는 현상으로 세포 내외에서 흔하게 일어날 수 있으며, 유전적 돌연변이, 단백질 번역오류, 열, pH, 산화, 이온세기 등에 따른 스트레스 그리고 불완전한 복합체 형성들을 포함한 다양한 사건들에 의해 발생할 수 있다고 한다.
본래 잘못 접혀서 올바르지 못한 구조를 갖는 단백질이 형성되면 샤페론(chaperone)에 의해 올바른 구조로 복구되거나 단백질 가수분해효소에 의해서 분해된다.
하지만, 일부 단백질은 잘못된 구조를 가졌음에도 불구하고 제거되지 못해 단백질의 응집체를 형성하기도 한다.
잘못 접힌 단백질은 무작위로 서로 결합하기 시작하고, 암, 심혈관 질환과 알츠하이머를 비롯한 다양한 병을 일으키는 원인이 된다.
그러므로, 3차원 단백질 구조 예측이 가능해지면 암, 알츠하이머 등의 여러 질병이 생기는 메커니즘을 더 잘 이해할 수 있으며, 그 밖에도, 정확한 단백질 구조를 아는 것은 백신 등의 치료제를 개발하는데 도움을 줄 수 있다.
대표적인 단백질 접힘 이상이 원인 질병 : 알츠하이머, 파킨슨 병, 헌틴텅 병(무도병), 루게릭 병, 암, 낭포성 섬유증, 2형 당뇨, 그 외의 300종 이상으로 예측된다고 한다.
최근 COVID19가 미친 영향을 생각하면, 신속한 신약 개발의 중요성을 실감할 수 있을 것이다.
단백질은 구조가 쉽게 무너지는 특성을 가지고 있어 구조를 알아내는 것이 매우 어렵다.
전통적으로 극초단판 현미경, 핵자기공명, X선 결정술과 같은 실험 기법을 사용하여 단백질의 구조를 연구했고 총 10만 개의 단백질 구조를 알아냈다. 그러나, 관측 기간이 길면 수년의 시간이 걸릴 수 있고, 단백질 하나를 분석하는데 수천만 달러 또는 최대 수십만 달러의 많은 비용이 든다. 이는 특수한 팬데믹 상황에서 빠른 치료제 혹은 백신 개발이 매우 힘들다는 것을 의미한다.
2. Method
FLOW CHART

Alpha Fold flow chart 위 그림은 Alpha Fold의 전체적인 flow chart이다. 정리해 보면 아래와 같다.
- MSA DATA를 입력으로 받는다.
- Deep ResNet을 통해 3개의 output을 얻는다.
- 거리 예측 (Distance Distribution)
- 뒤틀림 예측 (Torsion Distribution)
- 예측한 거리 및 뒤틀림 각을 기반으로 potential을 계산
- Potential을 기반으로 단백질 구조를 예측
MSA feature extraction
Multiple Sequence Alignment (MSA)란 3개 이상의 DNA, RNA, Protein과 같은 서열들을 sequence alignment를 진행하는 것을 말한다.
다수의 서열 (주로 FASTA 형태)의 공통된 부분을 찾아 정렬한다.
그럼 MSA 구조를 왜 사용하는 걸까?
우선 Folding 된 구조에서 단백질은 접점이 있다는 특징을 가지고 있다.

https://www.youtube.com/watch?v=MHb18hhKQsY 단백질의 접점을 우리가 먼저 알아낸다면 문제를 더 쉽게 만드는 것이 가능하기 때문에 이는 이전의 많은 연구에서 사용되던 방식이다.
접점을 찾는 방법은?

https://www.youtube.com/watch?v=MHb18hhKQsY 아미노산은 변형이 생기게 되면 보통 접점으로 접해있는 부분이 같이 변형을 이루게 되는데 이를 공진화라고 한다.
즉, 공직화가 이루어진 곳을 찾으면 우리는 접점을 찾아낼 수 있을 것이다.

https://www.youtube.com/watch?v=MHb18hhKQsY DNA는 진화를 이루며 변형되어 간다. 종들 간의 유전자를 비교하여 변형된 부분을 찾아내게 된다.
유전자를 비교하려면 유전자를 서로 정렬해서 비교해야 한다. 우리는 이 정렬한 과정을 MSA라고 부른다.
MSA를 통해서 특정 부분의 아미노산이 공진화한 것을 발견한다면 우리는 그 부분이 접점이구나! 하고 추정할 수 있을 것이다.
Deep ResNet

AlphaFold의 deep neural network는 단백질의 뒤틀림 각 및 residue 사이의 거리를 예측하는 역할을 담당한다.
단백질의 전체 길이가 L이라고 하였을 때 L x L의 distogram의 일부 64 x 64 영역에 대해서 residue 간의 거리를 예측한다.

총 220개의 residual convolution block을 사용하는 ResNet의 구조를 가지고 있다.
각 Residual block은 (Bathch norm, ELU)를 각각 가지고 있으며 project down, dilated conv, project up을 각각 사용하고 있다.
Dilated convolution을 사용하여 더 넓은 receptive field를 가질 수 있게 함으로 아미노산에 대한 정보가 주변 영역으로 확산되도록 하였다고 한다.
Distogram (Distance Distribution)

기존에 방법들은 residue 간의 거리를 그냥 측정하였지만 AlphaFold는 이 거리의 확률분포를 구하게 된다.
위의 그림에서 feature map은 각 필셀이 확률분포를 나타내고 있는 것이다.
DeepMind에서는 이것을 distogram이라고 얘기하고 있다.
Folding Distribution
folding 구조를 수학적으로 표현하기 위해서 간단하게 아미노산 중심 원자들의 3차원 좌표를 생각할 수 있을 것이다.

https://www.youtube.com/watch?v=MHb18hhKQsY AlphaFold에서는 뒤트림 각도를 통해서 folding 구조를 표현한다.

https://www.youtube.com/watch?v=MHb18hhKQsY 단백질이 folding 되더라고 아미노산의 구성에서는 변화가 없다. 비틀림 각도만 변하게 될 뿐이다. 이 비틀림 각도의 확률 분포를 구하는 방식으로 학습을 진행하게 된다.
Potential
실제 세계의 단백질은 potential이 가장 낮아지는 구조로 접히게 된다.
그 때문에 전 단계에서 계산된 potential을 일종의 loss function으로 생각할 수 있다.
그럼 potential function을 만들어야 할 것이다.
potential function은 distogram과 뒤틀림 각도의 확률 분포를 통해서 구할 수 있다.
Distance potential : (Initialization structure) x (Distogram)
Torsion potential : (Initialization structure) x (torsion distribution)
x : cross-product
마지막으로 물리적 제약을 고려한다.
AlphaFold의 경우 중심 원자들의 구조를 먼저 예측한다. 예측된 중심원자에 residue를 붙이게 되는데 이때 residue 끼리 부딪히는 steric clash가 발생할 수 있다. 이를 계산하기 위한 potential function을 포함하고 있다.
최종적으로는 아래와 같은 Potential Function을 가지게 된다.
score2_smooth function paper : Macromolecular modeling with Rosetta.
V_total (ϕ, ψ) = V_distance (ϕ, ψ) + V_torsion (ϕ, ψ) + V_(socre2_smooth) (ϕ, ψ)Potential을 구한 뒤 학습과정 정리
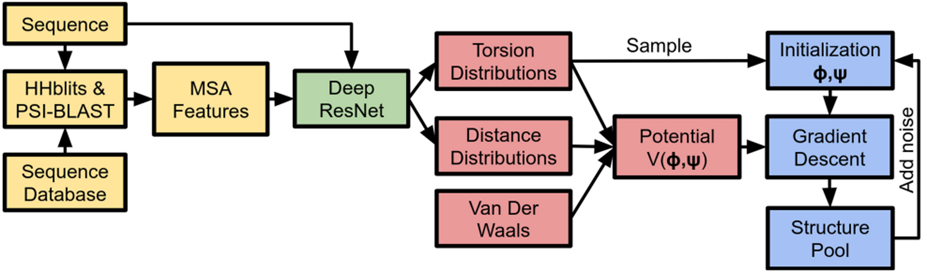
- Deep ResNet에서 구한 뒤트림 각도의 예측값으로 최조의 단백질 folding 구조를 제안
- 앞서 구한 potential function을 이용해서 해당 구조의 potential 값을 구한다.
- Gradient Descent를 진행
- Structure Pool에 잠정적 정답을 저장한다.
- Initialization 된 것에만 치중하여 학습하면 Local minima에 빠질 수 있으므로 이를 방지하기 위해 noise를 추가해 준다.
- 노이즈가 추가된 시점부터 시작해서 반복해서 gradient descent를 진행하고 해당 iteration의 최적값을 예상 답으로 저장한다.
- 최종정으로 potential 이 가장 낮은 구조를 선택한다.
3. Experiment


AlphaFold를 통해 제약 분야에서 새로운 붐이 일어났다고 한다.
2022년에 공개된 AlphaFold2는 앞도적인 성능으로 대회에서 우승을 하였다 한다. 이를 통해 이제까지 정복하지 못했던 질병을 인류가 정복하는 것에 한 발자국 더 다가섰다고 생각한다.
앞으로 인공지능을 통한 제약분야의 무한한 발전을 확인할 수 있는 계기가 되었다.
생명공학, 바이오 공학, 제약에 대해서 자세히 모르기 때문에 이해하는 것에 어려움이 있었지만 개인적으로 AI를 공부하고 연구하는 사람으로서 이 분야에 대해서 앞으로 계속 공부해야 할 이유를 크게 느끼게 되는 논문이었다.
Reference
https://contents.premium.naver.com/hughkim/knowledge/contents/211224015523280YD
단백질 잘못 접힘, 아밀로이드성 응집, 그리고 질병
4장 단백질 잘못 접힘, 아밀로이드성 응집, 그리고 질병 고려대학교 화학과 김준곤, 임동준 단백질의 접힘(protein folding)에 의해 만들어진 고유한 구조는 단백질의 기능과 직결됩니다. 그래서 단
contents.premium.naver.com
https://www.ksmcb.or.kr/abst/file/2011_17b.pdfMultiple Sequence Alignment의 원리 및 응용
안녕하세요. 세니젠입니다^^오늘은 Multiple Sequence Alignment의 원리 및 응용에 대해 알아보도록 하...
blog.naver.com
AlphaFold: 딥러닝으로 단백질 구조 예측하기
김성준, Virtual Machine & Optimization Lab in Seoul Nat’l University
medium.com
[기술 소개] AlphaFold : 단백질의 3D 구조를 AI가 예측한다 (한글)
쓰이지 않고 존재만 하는 기술이 무슨 의미가 있을까요? AI가 생명공학 분야에서 멋지게 활약한 사례인, AlphaFold를 소개합니다.
velog.io
'AI > Bio & Medical' 카테고리의 다른 글